
前言
在活细胞中,蛋白质之间的动态相互作用被认为在调节许多信号转导途径中起关键作用,并有助于广泛的其他关键过程。过去,阐明这种相互作用机制的经典生物化学方法是司空见惯的,但是在自然细胞环境中可能发生的弱或短暂的相互作用通常对这些技术是透明的。例如,在固定细胞中使用免疫荧光显微镜共定位可疑蛋白质伴侣已成为原位检查相互作用的常用方法,并且已经基于该技术提出了许多文献报告。然而,由于荧光显微镜的分辨率比典型蛋白质的大小小几百倍,因此共定位通常会导致有问题的结果。一个很好的类比是,荧光显微镜产生的信息相当于两个学生在一个大讲堂里的知识。它没有提供必要的分辨率来确定学生是否在同一间教室,或者更好的是,他们是否坐在相邻的桌子上。
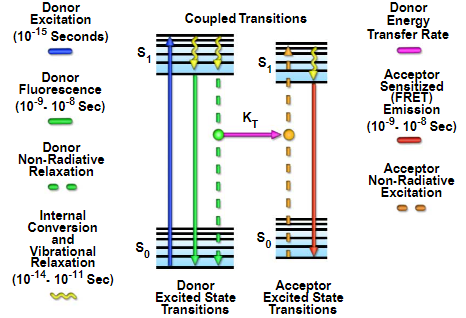
图1-福斯特共振能量转移雅布隆斯基图
典型的荧光显微镜技术依赖于荧光团在一个波长(激发)处吸收光,然后以更长波长发射二次荧光。激发和发射波长通常相隔数十至数百纳米。用特定的荧光团标记细胞成分,如细胞核、线粒体、细胞骨架、高尔基体和膜,使其能够在固定制剂和活制剂中定位。通过同时用具有分离激发和发射光谱的单个荧光团标记多个亚细胞结构,可以采用专门的荧光滤光片组合来检查单个细胞或组织切片内标记分子的接近程度。使用这种技术,比光学分辨率极限更近的分子似乎是重合的(并且据说是共定位的)。这种明显的空间接近意味着分子关联是可能的。然而,在大多数情况下,正常的衍射极限荧光显微镜分辨率不足以确定生物分子之间是否确实发生了相互作用。
共定位测量充其量是提示性的,最坏的情况是误导性的,特别是考虑到许多信号通路使用相同的细胞结构,例如,用于许多受体复合物内化的网格蛋白包被的凹坑。两个分子或蛋白质实际上是相邻的,而不仅仅是存在于同一个邻域的知识,为它们的相互作用潜力提供了更可靠的确定。历史悠久的电子显微镜技术具有足够的分辨率来满足高精度定位的需求,但缺乏产生可靠结果所需的精确标记方法。此外,许多共定位技术通常用于固定细胞内,这排除了通过活细胞测定获得的非常理想的动态测量。使用多色荧光蛋白进行荧光成像很容易允许对活细胞进行实验,这对于瞬态相互作用的测定是必要的,但该方法的空间分辨率相对较差,仅限于约200纳米。
通过应用Förster(或荧光)共振能量转移(FRET)显微镜技术可以克服确定蛋白质分子空间邻近性的局限性。FRET仅在两个适当定位的荧光团之间发生,当它们之间的距离为8至10纳米或更小时。因此,FRET非常适合研究位于彼此几纳米内的两个分子之间发生的蛋白质相互作用。在过去的十年中,由于需要使用与绿色荧光蛋白(GFP)及其突变衍生物融合来遗传靶向特定蛋白质和肽的应用的增加,FRET方法越来越受欢迎。两种光谱不同的荧光蛋白(称为FP-FRET)之间的FRET已广泛用于两种截然不同的实验技术,如下所述。图1所示是Jablonski能量图,说明了FRET中供体发射和受体吸光度之间涉及的耦合激发态转变。吸收和发射跃迁由垂直直箭头(蓝色、绿色和红色)表示,而振动松弛由波浪状黄色箭头表示。耦合跃迁用虚线绘制,表明它们在Jablonski图中的正确位置,如果它们是由光子介导的电子跃迁引起的。在合适的受体存在下,供体荧光团可以将激发态能量直接转移到受体上,而不会发射光子(图1中的紫色箭头所示)。所得荧光敏化发射具有与受体的发射光谱相似的特性。
在活细胞中广泛实施FRET研究的主要障碍之一是缺乏用适当的荧光团标记特定细胞内蛋白质的合适方法。最近开发的具有广泛光谱谱的荧光蛋白和蛋白质嵌合体(融合以及生物传感器)的日益复杂,导致了许多潜在的荧光蛋白对,这些荧光蛋白对在FRET实验中很有用。荧光蛋白在FRET中的应用涉及将选定的一对整合到生物传感器(单个基因编码结构)中,或者在两种单独的蛋白质之间进行分子间测量,每种蛋白质都融合到不同的荧光蛋白上。后一种方法已被用于对各种蛋白质相互作用进行成像,包括受体的寡聚化和阐明转录因子的功能。然而,对独立表达的蛋白质嵌合体进行FRET测定要困难得多,因为当单独的荧光实体在活细胞中表达时不可避免地会发生化学计量变化。无论难度如何,当安装适当的控制装置并以精确的精度进行调查时,这种性质的实验都可以产生信息丰富的结果。
荧光蛋白生物传感器
荧光蛋白生物传感器在报告各种细胞内过程方面发现了广泛的用途。通过将荧光蛋白对创造性地融合到执行生理信号传导各个方面的关键功能的生物聚合物中,研究科学家开发了许多新的分子探针,可用于重要过程的光学活细胞成像,如钙波诱导、环核苷酸信使效应、pH 变化、膜电位波动、磷酸化和细胞内蛋白酶作用。生物传感器构建的另一种但非常有用的策略涉及修改荧光蛋白骨架结构本身,要么将肽分解成单独的单元,这些单元在体内组合以产生荧光(一种称为双分子F荧光C修饰的技术;BiFC)或将天然氨基和羧基末端连接在一起,并在分子内创建传感器肽的插入位点。
第一个荧光蛋白生物传感器是一种名为Cameleon的钙指示剂,通过将蛋白质钙调蛋白和肌球蛋白轻链激酶(M13结构域)的钙钙调蛋白结合结构域夹在增强的蓝色和绿色荧光蛋白(EBFP和EGFP)之间构建。在细胞内钙水平增加的情况下,M13结构域结合钙调蛋白肽以产生荧光蛋白之间的FRET增加。不幸的是,该传感器受到非常低的动态范围(荧光增加1.6倍)的阻碍,并且由于EBFP的亮度不足和光稳定性差而难以可视化。使用相同模板的改进版本结合了青色和黄色变体ECFP和EYFP,以产生更高的信号电平,并且当通过插入钙敏感肽在第七个β开始时产生YFP衍生物(称为camgaroos)时,获得了更好的结果-在荧光蛋白骨架中链。位于这种不寻常位置的传感器肽在保持高水平的荧光方面耐受性非常好。另一种策略利用荧光蛋白中常见的独特桶结构,通过连接天然N和C末端并在结构中心区域内的几个位置之一(通常在环中)创建新的起始密码子来重新配置蛋白质的末端。这些结构修饰的衍生物被称为环状排列荧光蛋白,可以与钙调蛋白和M13融合,以产生出色的钙生物传感器。
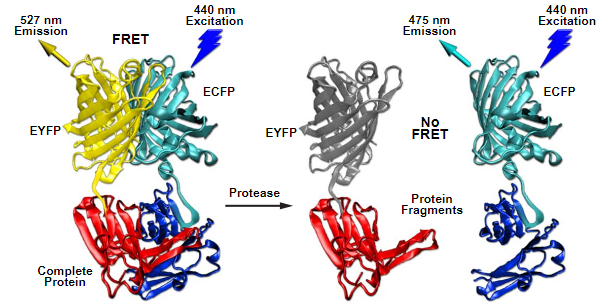
图 2-用于蛋白酶活性的荧光蛋白 FRET 生物传感器
钙生物传感器之后很快出现了pH、磷酸化和蛋白酶活性的遗传指标。可以使用两种通用方法来调整荧光蛋白作为pH传感器。第一种依赖于EGFP(pKa = 5.9)和EYFP(pKa = 6.5)对酸性环境的荧光敏感性,以及其他蛋白质的相对不敏感性,例如ECFP(pKa = 4.7)或DsRed(pKa = 4.5)。EGFP或EYFP与灵敏度较低的荧光蛋白融合产生可用于测量细胞内区室酸度的比例探针。第二种方法依赖于天然(野生型)GFP的质子化变化,导致天然蛋白质的双峰光谱谱发生变化。一类名为pHluorins的探针,源自wtGFP,随着pH值的降低,激发峰从470纳米变为410纳米。还开发了双发射pH传感器,其在绿色和蓝色光谱区域中具有峰值。虽然无法实时报告激酶活性,但磷酸化生物传感器由含有来自特定激酶的磷酸化基序的肽和夹在两个具有FRET功能的荧光蛋白之间的磷酸肽的结合域组成。当生物传感器被激酶磷酸化时,磷酸肽结合域与磷酸化序列结合,从而调用或破坏FRET。事实证明,这种简单的策略可以产生强大且高度特异性的生物传感器。与许多其他生物传感器一样,主要缺点是动态范围减小。
也许用于筛选新的或改进的FRET对的最广泛使用的生物传感器设计涉及蛋白酶切割测定(见图2)。简单的基序由两个荧光蛋白组成,通过含有共识蛋白酶切割位点的短肽连接在一起。通常,传感器表现出非常强的能量传递,在接头序列裂解时完全消除。由于该技术通常具有高动态范围水平,因此可用于筛选具有黄色,橙色和红色受体的新青色和绿色FRET供体。最大的蛋白酶生物传感器系列包含一个对半胱天冬酶家族蛋白酶之一敏感的切割位点,这使得传感器能够在诱导细胞凋亡期间进行检查。在过去的几年中,已经报道了大量使用敏化荧光蛋白和FRET对的新型生物传感器。尽管使用ECFP和EYFP衍生产品的FRET传感器的动态范围持续受到限制,但这种策略已被广泛采用,这可能是由于比率测量的简单性和探头结构的便利性。毫无疑问,使用更先进的荧光蛋白组合会出现新的策略,这些组合有助于增加这种非常有用的探针的动态范围和其他特性。
FRET的基本原则
FRET的基本机制涉及处于激发电子状态的供体荧光团,其可以通过长程偶极子 - 偶极相互作用以非辐射方式将其激发能量转移到附近的受体荧光团(或发色团)。支持能量转移的理论基于将激发的荧光团视为振荡偶极子的概念,该偶极子可以与具有相似共振频率的第二个偶极子进行能量交换。在这方面,共振能量传输类似于耦合振荡器的行为,例如一对以相同频率振动的音叉或无线电天线。相比之下,辐射能转移需要光子的发射和重吸收,并且取决于样品的物理尺寸和光学特性,以及容器和波前路径的几何形状。与辐射机制不同,共振能量转移可以产生大量关于供体-受体对的结构信息。
共振能量转移对荧光团周围的溶剂壳不敏感,因此会产生与溶剂依赖性事件(如荧光猝灭、激发态反应、溶剂弛豫或各向异性测量)所揭示的分子信息所揭示的分子信息不同的分子信息。溶剂对参与共振能量转移的荧光团的主要影响是对供体和受体的光谱特性的影响。非辐射能量转移发生在比短程溶剂效应更长的距离上,并且位于相关荧光团之间的成分(溶剂和宿主大分子)的介电性质对共振能量转移的功效影响很小,这主要取决于供体和受体荧光团之间的距离。
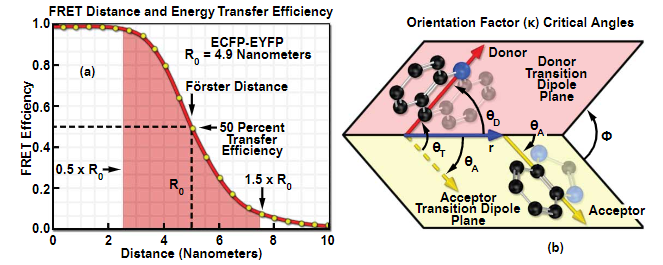
图 3 - FRET中的福斯特距离和方向因子变量
FRET现象不是由光子发射介导的,而且,甚至不需要受体发色团是荧光的。然而,在大多数应用中,供体和受体都是荧光的,并且能量转移的发生表现为供体荧光的猝灭和荧光寿命的缩短,同时也伴随着受体荧光发射的增加。共振能量转移理论最初是由西奥多·福斯特(Theodor Förster)提出的,为了纪念他的贡献,最近以他的名字命名。Förster理论表明,FRET效率(E)随两个分子之间距离的六次方的反六次方而变化(用r表示):
EFRET= 1/[1 + (r/R0)6]
其中R(0)是FRET效率为50%的特征距离,可以针对任何一对荧光分子计算(该变量也称为Förster半径,下面将更详细地讨论)。理论荧光团对(增强型青色和黄色荧光蛋白)的FRET效率如图3(a)所示。由于对两个分子之间的距离(r)的六次幂成反比,曲线急剧下降。对于小于R(0)的距离,FRET效率接近最大值,而对于大于R(0)的距离,效率迅速接近零。测量FRET的有用范围由图3(a)中的红色阴影区域表示,其限制为0.5和1.5 xR(0)。FRET可以有效地用作接近R(0)的距离的分子标尺,事实上,FRET已经通过使用精密光谱方法适用于结构生物学中的此类目的。然而,对于细胞生物学中的大多数应用,可用的信噪比将FRET实验限制为更二进制的读数。实际上,测量通常只能区分高FRET和低FRET,或者简单地区分FRET的存在和不存在。
如前所述,R(0)可以很容易地计算出任何一对荧光分子。水溶液(或缓冲溶液)中R(0)的值由一个相当简单的方程确定,该方程具有完善的输入参数:
R0= [2.8 x 1017× Κ2× QD× ΕA× J(λ)]1/6纳米
其中Κ(2) 或kappa 平方表示两个荧光团偶极子之间的取向因子(用于计算取向因子的角度摘要,参见图 3(b)),Q(D) 是供体量子产率,Ε(A) 是以每厘米倒数摩尔为单位的最大受体消光系数,J(λ) 是光谱重叠积分(见图 4)在归一化供体荧光F(D)(λ)和受体激发光谱E(A)(λ)之间,根据公式:
J(λ) =∫FD(λ) × EA(λ)×λ4dλ
虽然数学可能看起来很复杂,但大多数参数都是常数,很容易在文献中找到。通常需要进一步解释的两个最重要的项是Κ(2) 和J(λ),即重叠积分。方向角变量(Κ(2))只是表明FRET耦合取决于两个荧光团之间的角度,其方式与无线电天线的位置影响其接收的方式大致相同。如果供体和受体彼此平行排列,则FRET效率将高于垂直定向。这种对齐程度定义了Κ(2)。虽然Κ(2) 可以在 0 和 4 之间变化,但通常假定它是 2/3,这是在所有可能角度上积分的平均值。对于几乎任何现实情况,Κ(2)接近2/3,研究人员通常无法调整该值(尽管有些人将荧光蛋白刚性地附着在感兴趣的靶蛋白上,这可能会导致显着的效果)。重叠积分J(λ)是两个光谱之间的重叠区域,如图4所示。可能影响FRET的其他参数是供体的量子产率和受体的消光系数。因此,为了最大化FRET信号,研究人员必须选择最高的量子产率供体,最高的吸收受体,以及光谱分布显着重叠的荧光团。该理论已被实验反复验证,并且没有其他机制可以使非对齐荧光探针的FRET最大化。
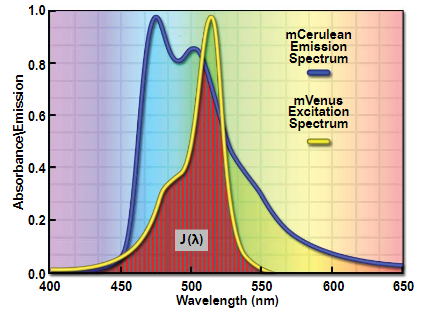
图4-激发和发射光谱重叠积分
应该注意的是,上面讨论的每个参数仅影响六次方的福斯特半径计算。因此,供体量子产率的加倍只会导致R(0)的变化为12.5%。由于FRET成像实验中使用的几乎所有荧光团都具有高量子产率(大于0.5)和消光系数(大于50,000),因此可能的Förster半径值范围限制在4到6纳米之间,并且大多数FRET对的平均值为R(0)~5纳米。鉴于FRET效率在很大程度上取决于FRET对之间的距离以及荧光团的相对取向,FRET可用于检测由两种蛋白质之间的亲和力变化或其结合构象的变化引起的蛋白质 - 蛋白质相互作用的变化。值得重复的是,对于细胞生物学中的大多数FRET成像应用,实验通常只区分两种状态(FRET和无FRET),并且需要额外的信息来帮助对观察到的FRET变化进行分子解释。
影响FRET测量的因素
在实践中,广泛的问题可能会使FRET测量复杂化和/或妥协,最终导致模棱两可或无意义的结果。主要问题之一是供体和受体荧光团在一起成像时可能表现出显着不同的亮度水平。虽然从理论上讲,这种差异应该不是问题,但在实践中,由于大多数仪器只能测量有限的动态范围,双荧光团成像可能导致一个通道饱和(对于较亮的荧光团),而另一个通道则由系统噪声主导(对于较暗的荧光团)。因此,只要有可能,最好使用亮度相当的供体和受体。
另一个可能限制FRET检测的因素是供体到受体的化学计量,其范围在10:1和1:10之间。在蛋白质 - 蛋白质相互作用的FRET测量中,这一因素可能是一个严重的限制,其中一方可能浓度过高。主要问题是在不进行FRET的荧光标记的背景下测量少量FRET的量。由于实际上没有什么可以改善这种情况的,因此属于这一类别的许多可能的蛋白质 - 蛋白质相互作用实验根本不适合通过FRET技术进行检查。对于上述仅由单个供体和受体构建的荧光蛋白生物传感器,化学计量是固定的,保证为1:1;因此,无论生物传感器浓度如何,这个问题都不会出现,信号电平保持不变。
渗漏(也称为串扰和交叉)的存在以及光谱重叠荧光团之间的交叉激发也是阻碍FRET研究的重要问题(见图5)。在某些情况下,受体可以直接用选择激发供体的波长区域的光激发(图5(a))。此外,来自供体的荧光可能会泄漏到受体荧光的检测通道中,特别是当供体和受体的发射光谱图表现出显着重叠时(图5(b))。由于这两个串扰源来自有机荧光团的光物理学,并且肯定会存在于任何FRET对中,因此在测量FRET时必须解决它们。选择光谱分离良好的荧光团是减少串扰的极好机制。然而,在大多数情况下,增加的频谱分离也会降低重叠积分(J(λ)),这在实践中通常意味着检测FRET信号的能力降低。
最后,如果两个荧光团未正确对齐(例如,Κ(2)值近似于零),或者它们根本不位于Förster半径内(大于6纳米),则可以降低FRET信号的水平。例如,如果两个标记的蛋白质相互作用,但荧光标记位于复合物的相对两侧,那么即使感兴趣的蛋白质被结合,也可能没有可检测到的FRET信号。在一般实践中,这种类型的假阴性很常见,尤其是对于荧光蛋白FRET伴侣。通常,在检测到足够可靠的FRET信号之前,需要几种标记策略。然而,在制作载体构建体或进行合成标记实验之前,通过明智地选择要使用的荧光团对,可以缓解(或部分缓解)上述每个问题。
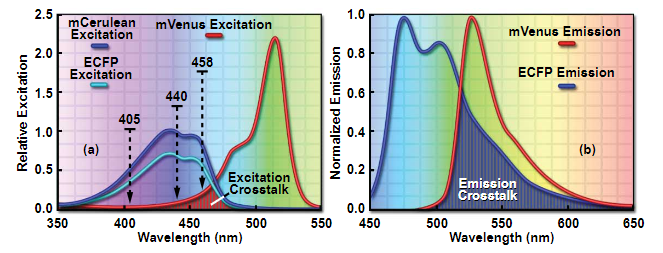
图 5- CFP-YFP 品对中的光谱渗漏(串扰)
图5所示是ECFP和mVenus的激发和发射光谱图的重叠,目前是FRET研究中最受欢迎的荧光蛋白对之一。这两种蛋白质在激发(图5(a))和发射(图5(b))光谱中都表现出相当大的重叠。FRET受体(mVenus;红色曲线)的直接激发可能很大,具体取决于用于激发供体的波长(ECFP;青色曲线或mCerulean;蓝色曲线),因为黄色蛋白质的消光系数高于青色蛋白质。当ECFP用作供体时,这种重叠尤其成问题,并且可以通过使用具有高消光系数的CFP变体(例如mCerulean)来部分抵消。请注意,图5(a)中的激发曲线按比例绘制,以反映黄色和青色蛋白质之间的消光系数差异。458纳米的激发产生的mVenus激发串扰水平比405或440纳米的激发高得多。ECFP的宽荧光发射光谱(图5(b))在整个mVenus发射区域表现出相当大的强度重叠。
FRET技术在细胞生物学应用中的应用
使用荧光蛋白生物传感器的研究者,或试图匹配融合到分离相互作用靶标的荧光探针的化学计量,应使用尽可能多的不同FRET分析技术来建立给定实验的方法。这种努力是有道理的,因为每个荧光蛋白FRET对都表现出独特的病理学,使其使用复杂化,需要清楚地了解用于测量大多数FRET测定中产生的相对较小的信号差异的光学显微镜参数。一旦系统和可能的结果建立得很好,那么最简单的方法就可以用于正在进行的程序。已经开发的用于成像FRET的技术列表非常广泛。一般来说,所有现有的测量FRET的策略都可以应用于荧光蛋白实验,但基于实际考虑,五种通用方法已被证明特别有用:
- 敏化发射 - 使用校正激发和发射串扰的算法进行双通道成像
- 受体光漂白 - 也称为供体去猝灭,该技术测量受体光漂白时供体发射的增加
- 荧光寿命成像显微镜(FLIM)- 荧光蛋白(或其他荧光团)供体寿命测量变化
- 光谱成像 - 在一个或两个波长下激发并测量供体和受体的完整光谱分布
- 荧光偏振成像 - 以高信噪比测量平行和垂直于激发的偏振
上面列出的每种FRET方法都有优点和缺点。例如,一方面,双通道成像是最简单的方法,但需要最复杂的一组控件。另一方面,FLIM可以明确测量FRET效率,并且仪器可用于集成到尼康A1 HD25 / A1R HD25共聚焦系统中。
敏化发射
通常也称为带对照的双色比成像,敏化发射可能是最简单的成像FRET。供体荧光团由特定波长(在宽场或共聚焦显微镜中)激发,并使用为供体荧光和受体荧光选择的发射滤光片收集信号。在两个荧光团的激发和荧光之间(不切实际的)不存在串扰的情况下,敏化发射将是一种完美的方法。然而,荧光蛋白之间的串扰是一个重大问题,通常需要广泛的对照实验来确定FRET的存在与否。因此,使用这种方法很难获得定量准确的FRET数据。在许多实验室中可用的宽场荧光显微镜上配置敏化发射相对简单,但必要的控制实验需要大量的图像处理来减去串扰分量,这显着增加了测量中的噪声水平和不确定性。
已经为敏化发射FRET成像开发了多种纠正方法。基本概念涉及使用不同的过滤器组合,其中包含多个样品:仅供体,仅受体,以及具有供体和受体的推定FRET样品。这些样品的发射值允许研究人员确定激励和发射通道中的预期串扰量,并将其从FRET测量中减去。从理论上讲,这种方法效果很好,但不幸的是,对图像处理的要求增加了所有图像中的噪声水平。因此,如果FRET信号很小,那么使用这种方法可能很难测量FRET。
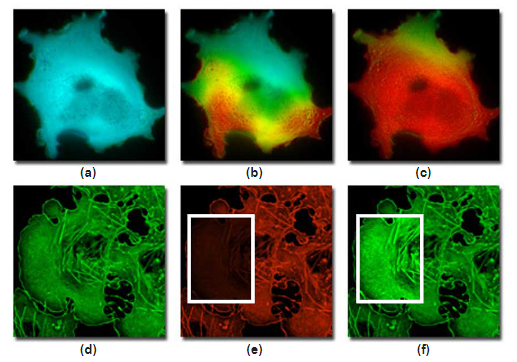
图 6- 敏化发射和受体光漂白 FRET(光漂白 FRET)
尽管存在上述困难,但敏化发射测量对于快速动态实验非常有用,其中FRET信号很大,因为能够同时获取两个图像。在检查荧光蛋白生物传感器时,敏化发射是一种特别有吸引力的技术,其中FRET动态范围很大,供体和受体的化学计量固定在1:1的比例。图2所示的蛋白酶生物传感器就是一个很好的例子。该嵌合体被设计为具有高FRET效率,当肽接头被酶切时,该效率基本上降至零。结果是一个大且易于测量的FRET变化,表明在活细胞内的给定时间和区域具有特定的蛋白酶活性。
受体光漂白
虽然仅限于一次测量,但受体光漂白(或供体去猝灭)也是一种简单的技术,通常会产生出色的结果。基本概念利用了供体荧光在FRET过程中被淬灭的事实,因为一些供体荧光能量被引导到受体。光漂白受体荧光团不可逆地消除了猝灭效应并增加了供体荧光的水平。如果FRET发生在荧光团之间,则当受体被移除时,供体荧光必须增加。通常,重要的是要确保受体光漂白不会降低供体荧光,并且受体的光漂白到其初始值的约10%。激光扫描共聚焦显微镜可以轻松满足这两个限制,但配备专用照明系统的宽视场或转盘显微镜也可以满足这两个限制。
受体光漂白技术的优点是非常直接、定量的,并且仅使用单个样品即可进行。FRET效率可以通过在光漂白受体后从受体强度中减去受体存在下的供体强度,然后将该值归一化为漂白后的供体强度来计算。主要缺点是受体光漂白是破坏性的,每个细胞只能使用一次,将其应用限制在那些不涉及动态测量的实验中。此外,光漂白是一个相对缓慢的过程,通常需要几分钟或更长时间。然而,无论使用哪种方法来测定FRET,在实验结束时进行受体光漂白测量几乎总是值得的。
图6所示是使用活细胞成像的敏化发射和受体光漂白FRET测定的示例。图6(a)显示了人宫颈癌上皮细胞(HeLa系),该细胞表达由mCerulean和mVenus组成的骆驼生物传感器,与含有钙调蛋白和M13结构域(如上所述)的中间钙敏感肽融合在一起。在添加钙诱导剂(离子霉素)之前,用440纳米照明激发细胞产生青色荧光,表明青色和黄色荧光蛋白之间缺乏FRET(图6(a))。加入离粒霉素后,延时双色比成像(敏化发射)记录钙波穿过细胞质,因为生物传感器响应荧光蛋白之间FRET水平的增加(图6(b)和6(c);FRET是假色的黄红色)。图6(d)-(f)中的非洲绿猴肾细胞(COS-7系)用合成花青染料Cy3(图6(d);绿色)和Cy5(图6(e);红色)标记,与霍乱毒素B亚基结合并靶向质膜。在膜内,两种染料的紧密接近会产生高水平的FRET。当仅在供体通道中观察荧光时,在细胞的选定区域(图6(e)中的白框)中的光漂白Cy5会增加相应区域中的供体淬灭(图6(f)中的绿色荧光增加)。
荧光寿命成像显微镜 (FLIM)
寿命测量是迄今为止确定 FRET(弗雷特)最严格的方法;此外,由于仅监测供体荧光,它们也不易产生串扰伪影。所有荧光分子在纳秒时间尺度上的荧光都表现出指数衰减,并且这种衰减的速率对猝灭荧光的环境变量很敏感。因此,FLIM的基本概念与受体光漂白的基本概念有些相关。供体荧光通过FRET相互作用猝灭,并且可以通过测量供体在FRET存在下的荧光衰减时间的减少来确定猝灭量。通过这种方式,FLIM提供了FRET效率的明确值。FLIM-FRET组合测量的优点之一是它们对直接受体激发伪影不敏感,以及荧光供体可以耦合到本身不是荧光的受体。这两个方面都有助于扩大研究人员可用的有用荧光蛋白FRET对的数量。
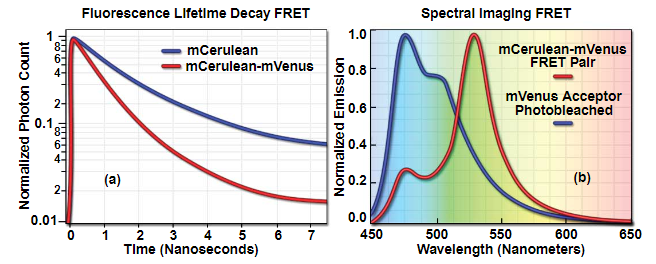
图 7 - FRET显微镜中的FLIM和光谱成像应用
FLIM有几个局限性,使其无法成为FRET成像的主要方法。首先,纳秒级生存区域的测量很复杂,仪器的获取和维护成本很高。此外,这种类型的复杂设备并不广泛可用。此外,FLIM通常是较慢的成像方法之一,可能需要几分钟才能获取每个图像,这限制了其在许多FRET实验中的实用性。随着制造商开发更加用户友好和更快的交钥匙商业系统,这些限制将来可能会被取消。另一个显着的缺点是活细胞中荧光蛋白的寿命通常显示多指数衰减,需要更全面的数据分析来进行定量FRET测定。此外,局部环境因素,如自发荧光或pH值变化,也会缩短测量的荧光寿命,导致伪影。因此,在解释活细胞中的FLIM-FRET数据时必须格外小心。
光谱成像
光谱成像技术是敏化发射FRET检测方法的变体,但不是通过两个单独的通道获取数据,而是在供体激发时收集包含供体和受体荧光的整个发射光谱。记录整个光谱是光谱学实验中的典型方法,但它是宽场和共聚焦显微镜工具选项板中相对较新的补充。该概念的中心前提是,收集整个荧光光谱不仅可以使用发射峰,还可以使用光谱尾的不同形状来分离重叠的光谱。在收集来自供体和受体荧光团的光谱时,可以确定供体和受体荧光的相对水平。
光谱成像技术需要专门的设备,但许多商用共聚焦显微镜上都有出色的系统,并且可以以较低的成本添加到传统的荧光显微镜上。对由于受体的直接激发或在共聚焦显微镜中使用两个激发波长而导致的串扰水平进行定量分析,可以准确确定FRET的量。这种方法的主要缺点是与获取完整频谱相关的信噪比降低,而不是通过基于滤波器的系统通过两个通道收集频谱。然而,随着越来越多的商业系统的开发和安装,光谱成像在FRET测定中的应用正在增加。在不久的将来,光谱成像很有可能成为进行FRET成像实验的主要方法之一。
图7(a)所示是伪FRET生物传感器的供体寿命衰减(mCerulean荧光蛋白)的变化,该生物传感器由mCerulean和mVenus荧光蛋白与10个氨基酸接头融合在一起。蓝色衰变曲线显示了在单独表达mCerulean的细胞中观察到的寿命,而红色衰变曲线显示了当细胞表达串联蛋白时获得的mCerulean寿命。请注意,当蛋白质参与共振能量转移时,mCerulean寿命会降低。曲线之间的面积表示在FRET对中通过FRET从mCerulean(供体)转移到mVenus(受体)的能量。图7(b)中的红色曲线描绘了同一伪生物传感器中450至650纳米的mCerulean-mVenus在活细胞中以405纳米激发时的发射曲线。从mCerulean到mVenus的能量转移在529纳米(mVenus发射最大值)处产生大量的发射峰,而在475纳米处(mCerulean的峰值发射波长)处的值要低得多(约25%)。在用514纳米激光对mVenus进行光漂白并重复光谱扫描后,在没有FRET伙伴的情况下,发射曲线转移到较低的波长,并且与mCerulean的光谱非常相似。这些光谱图在475和529纳米处的强度差异与偶联蛋白质之间的FRET效率有关。
偏振各向异性成像
荧光偏振的测量为荧光蛋白FRET的高对比度鉴别提供了特别的优势。该概念基于这样一个事实,即用偏振光激发选择一组荧光分子,其吸收矢量与激发光的偏振矢量平行排列。激发后,大部分荧光发射将保持平行于激发的偏振,因此荧光在偏振方面可以被认为是各向异性的。如果分子在纳秒荧光寿命期间旋转,则各向异性将消失。然而,由于荧光蛋白很大且旋转缓慢,因此它们的荧光在测量过程中不会在很大程度上去极化。如果FRET发生在两个略微错位的荧光蛋白之间,则偏振荧光发射将以不同的角度(从激发矢量)出现,这模拟了荧光蛋白的旋转。
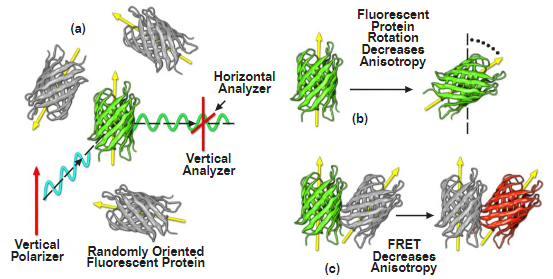
图 8- 偏振各向异性 FRET 成像
这种方法的主要优点是易于测量平行和垂直于激发矢量的荧光偏振,具有高信噪比。由于偏振各向异性数据可以快速采集,并且图像处理要求最低,因此该技术非常适合高内涵筛选应用。但是,必须避免直接激励受体,因为它会降低供体信号并降低测量的信噪比。此外,尽管这种技术在区分 FRE 的存在与不存在方面非常出色,但它并不是区分强 FRET 和弱 FRET 的好方法。最后,高数值孔径物镜的偏振可能会降低,因此偏振FRET实验应仅限于数值孔径为1.0或更小的物镜成像。
图8所示是使用荧光蛋白作为模型系统的偏振各向异性的图形说明。当随机取向的荧光蛋白群(图8(a))被线偏振光(青色波)激发时,只有那些吸收偶极向矢量平行于偏振方位角的分子被优先激发。正确定向荧光蛋白的发射可以使用与激发光偏振矢量(绿波)平行的分析仪作为信号进行观察。产生的各向异性是取向程度的指标,可以通过垂直和水平定向分析仪测量和比较发射强度来确定。如果荧光蛋白在实验的时间尺度上旋转(图8(b)),或者如果由于FRET将激发能转移到具有不同方向的相邻蛋白质(图8(c)),则各向异性信号水平将降低。如上所述,由于大荧光蛋白分子的共振能量转移比分子旋转发生得更快,因此FRET引起的去极化可以很容易地与旋转过程中发生的各向异性损失区分开来。
在FRET中使用荧光蛋白的注意事项
用于检查活细胞中FRET的合适探针的选择是有限的。合成荧光团是固定细胞共振能量转移研究的理想选择,在活细胞中难以给药和靶向。同样,量子点可用于标记膜组件以检查细胞外部的现象,但它们也无法穿透膜,因此在细胞内区室(如细胞核、线粒体或内质网)中几乎没有用处。基因编码的荧光蛋白目前代表了活细胞中FRET高分辨率成像的最佳候选者,每年在该领域发表的文献量证明了这一点。然而,使用合成荧光团和量子点测量FRET时遇到的许多典型伪影在应用于荧光蛋白时尤其严重。例如,与合成材料中发射光谱图的30-40纳米带宽相反,荧光蛋白中的发射光谱图范围约为60纳米至100纳米,在试图分离供体和受体荧光时通常会导致显着重叠。荧光蛋白的宽光谱也限制了在FRET和其他类型的成像实验中可以一起使用的探针数量。此外,荧光蛋白在亮度水平上表现出很大的变化。例如,最受欢迎的供体蛋白之一ECFP的亮度比其常见的黄色受体伴侣EYFP低五倍。
荧光蛋白发色团周围是一个220+氨基酸多肽,缠绕成一个三维圆柱形结构,尺寸约为2.4×4.2纳米(称为β桶或β-罐),由广泛的氢键多肽β-折叠组成,包围并保护含有发色团的中心α-螺旋(见图9).枪管的末端覆盖有半螺旋肽区域,用于阻止离子和小分子进入。蛋白质的内部被氨基酸侧链和水分子紧密地包裹着,以至于氧气、离子或其他侵入的小分子几乎没有扩散的空间,这些小分子设法穿过桶的末端。这些有利的结构参数是荧光蛋白弹性光稳定性和优异性能的部分原因,也有助于降低FRET效率。桶的大尺寸有效地用肽残基屏蔽相邻的荧光蛋白发色团(限制接近距离为2至3纳米;如图9中的红线所示),导致最大FRET效率降低到理论值的约40%。无论如何,使用荧光蛋白进行活细胞FRET成像的众多好处远远超过成本。

图 9-荧光蛋白结构特征
使荧光蛋白发生的高度光谱带宽重叠和大小问题更加复杂的是它们的寡聚化倾向。迄今为止发现的几乎所有荧光蛋白都显示出至少有限程度的四级结构,例如天然维多利亚绿荧光蛋白及其衍生物在高浓度下固定时二聚化的弱趋势。在珊瑚和海葵中分离的天然黄色、橙色和红色荧光蛋白的严格四聚化基序也证实了这种趋势。对于细胞生物学中的许多应用来说,寡聚化可能是一个重大问题,特别是在荧光蛋白与靶向特定亚细胞位置的宿主蛋白融合的情况下。一旦表达,嵌合体的荧光蛋白部分诱导的二聚体和高阶低聚物的形成可以产生非典型定位,破坏正常功能,干扰信号级联,或限制融合产物聚集在特定细胞器或细胞质内。当荧光蛋白与本身参与天然低聚物形成的伴侣融合时,这种效应尤其明显。具有仅形成弱二聚体的蛋白质的融合产物(实际上,大多数维多利亚风琴变种)可能不会表现出聚集或不正确的靶向,前提是局部浓度保持较低。然而,当弱二聚体荧光蛋白靶向特定的细胞区室(例如质膜)时,在某些情况下,局部蛋白质浓度可以变得足够高以允许二聚化。在进行分子间FRET实验时,这可能是一个特别值得关注的问题,这可能会产生复杂的数据集,有时会受到二聚伪影的影响。另一方面,在某些情况下,Aequorea蛋白中天然存在的弱二聚化可用于增加生物传感器中的FRET信号,否则这些传感器的动态范围将受到限制。
毒性是由于合成荧光团浓度过高以及定位不良的荧光蛋白的过度表达或聚集而引起的问题。此外,显微镜成像室中最佳标记的哺乳动物细胞的健康和寿命也可能受到许多其他有害因素的影响。其中最重要的是光诱导损伤(光毒性),当荧光标记的细胞反复暴露于激光和高强度电弧放电灯的照明下时。在激发状态下,荧光分子倾向于与分子氧反应产生自由基,从而破坏亚细胞成分并损害整个细胞。荧光蛋白由于其荧光团深埋在保护性多肽包膜内,通常对细胞没有光毒性。在设计FRET实验时,应选择具有最长激发波长的荧光蛋白组合,以尽量减少短波长照明对细胞的损害,特别是在长期成像实验中。因此,与其用蓝色或青色荧光蛋白(分别由紫外线和蓝色照明激发)制造聚变产物和生物传感器,不如在光谱的黄色、橙色和红色区域发射的变体将更加理想。
研究人员在使用新的荧光蛋白生物传感器和细胞系时应注意进行必要的对照实验,以确保细胞毒性和光毒性伪影不会掩盖FRET结果或其他重要的生物现象。在某些情况下,亲脂性试剂会引起有害效应,在瞬时转染后的细胞系成像过程中,这些效应可能与荧光蛋白毒性混淆。与单体水母蛋白相比,来自珊瑚的寡聚荧光蛋白(如上所述)形成聚集体(结合亚细胞定位不良)的倾向要大得多,但任何变体都可能发生折叠不当的融合产物。最近,据报道,一种能够在绿光照射下产生活性氧(ROS)的荧光蛋白是发色团辅助光灭活(CALI)使特定蛋白质失活的有效剂。这种基因编码的光敏剂被恰当地命名为KillerRed,能够在显微镜下照射时杀死细菌和真核细胞。先前对EGFP光毒性的研究表明,即使通过发色团能够产生单线态氧,荧光蛋白作为光敏剂的效率相对较低。然而,长时间照射表达EGFP及其变体的细胞会导致生理改变和最终的细胞死亡,这是长期成像实验中光毒性潜力的明确信号。
在活细胞实验中,荧光蛋白与合成荧光团相比,由于光漂白率降低,因此对于扩展成像非常有利。尽管荧光蛋白在光稳定性方面存在高度的不相关变异性,但大多数变体可用于短期成像(从1到25次捕获),而几种更光稳定性的蛋白质可以用于跨越24小时或更长时间的延时序列(其中收集了数百到数千张图像)。然而,对于每种照明场景(宽场、共聚焦、多光子、扫场等),都必须研究任何特定蛋白质的长期稳定性,因为当电弧放电灯与激光系统产生照明时,通常观察到相同蛋白质的光稳定性差异。因此,就光稳定性而言,荧光蛋白的选择取决于许多参数,包括照明条件、表达系统和成像设置的有效性。
潜在的荧光蛋白FRET合作伙伴
在过去的几年中,已经开发和改进了各种新的荧光蛋白变体,以具有跨越200纳米范围(从大约450纳米到650纳米)的发射曲线,从而填补了许多空白,在每个颜色类别中提供潜在有用的FRET合作伙伴。在蓝色(440纳米至470纳米)和青色(470纳米至500纳米)光谱区域开发蛋白质的最新进展已经产生了几种可用于成像和FRET研究的新探针。三个蛋白质工程小组报告了改进的蓝色Aequorea荧光蛋白变体,与EBFP相比,其亮度和光稳定性显着更高。这些蛋白质名为蓝铜矿,SBFP2(强烈增强的蓝色FP)和EBFP2(见表1),为蓝色光谱区域中活细胞的成功长期成像提供了第一个真正的希望,并且都具有与EGFP和衍生物结合在FRET生物传感器中的重要适用性。新的蓝色报告基因中最亮和最稳定的EBFP2在融合中表现出典型的GFP样行为,并且已被证明是绿色光谱类别蛋白质的出色FRET供体。所有蓝色荧光蛋白都可以使用标准DAPI滤光片组或售后制造商提供的专有BFP组在荧光显微镜中轻松成像。
青色光谱区域中的荧光蛋白在与黄色发光蛋白配对时已被广泛用作FRET供体,并且由原始AequoreaECFP的变体主导,直到引入单体蓝绿色报告基因,称为mTFP1。与AequoreaCFP相比,蓝绿色荧光蛋白表现出更高的亮度和酸稳定性,并且更具可光性。mTFP1的高发射量子产率(见表1)与黄色或橙色荧光蛋白结合时,作为FRET供体,是青色衍生物mECFP和mCerulean的极佳替代品。进一步的研究已经产生了青色光谱类别的有用蛋白质。在最近引入的改进的青色荧光蛋白中,CyPet和称为Cerulean的增强型青色变体作为融合标签,FRET生物传感器和多色成像的候选者显示出最大的前景。蔚蓝的亮度至少是ECFP的2倍,并且在FRET研究中,当与黄色发光荧光蛋白(如金星)结合使用时,已被证明可以显着提高对比度和信噪比。名为CyPet的CFP变体(来自首字母缩略词:Cy,荧光Protein,代表energytransfer)是通过利用荧光激活细胞分选(FACS)来优化FRET的青色和黄色配对的独特策略得出的。CyPet的亮度大约是EGFP的一半,是Cerulean的三分之二,但在37摄氏度时表现相对较差。然而,与CFP相比,CyPet具有更多的蓝移和更窄的荧光发射峰,这大大增加了其多色成像的潜力。
将有益的折叠突变引入ECFP的单体变体中,产生了具有增强亮度,折叠效率,溶解性和FRET性能的新变体。被称为超级CFPs(SCFPs),新的报告基因在细菌中表达时比亲本蛋白更亮,在哺乳动物细胞中几乎亮两倍。这些高性能探头应该既可用于常规融合标签,也可用于创建具有高动态范围的新型CFP-YFP FRET生物传感器。另一种新的单体青色报告基因TagCFP来源于水母Aequorea macrodactyla的GFP样蛋白。关于该蛋白质的具体细节在文献中不可用,但它可以作为哺乳动物克隆载体和Evrogen的融合物进行商业购买。据报道,TagCFP比ECFP和Cerulean更亮,但具有相似的耐酸性。另一种青色发射蛋白Midoriishi-Cyan(缩写为MiCy)最初被设计为与单体Kusabira Orange(mKO;见表1)的新FRET组合中的供体,以产生具有高光谱重叠的生物传感器(Förster距离为5.3)。这种蛋白质具有青色光谱区域任何探针报告的最长吸收和发射波长曲线(分别为472和495纳米)。MiCy表现出的高摩尔消光系数和量子产率使蛋白质具有与蔚蓝相同的亮度。
表1-所选荧光蛋白FRET对的特性
| 蛋白质对 | 供体激发最大值 (nm) |
受体发射最大值 (nm) |
供体量子产量 | 受体摩尔消光系数 | 福斯特距离 (海里) |
亮度比 |
| EBFP2-mEGFP | 383 | 507 | 0.56 | 57,500 | 4.8 | 1:2 |
| ECFP-EYFP | 440 | 527 | 0.40 | 83,400 | 4.9 | 1:4 |
| Cerulean-Venus | 440 | 528 | 0.62 | 92,200 | 5.4 | 1:2 |
| MiCy-mKO | 472 | 559 | 0.90 | 51,600 | 5.3 | 1:2 |
| TFP1-mVenus | 492 | 528 | 0.85 | 92,200 | 5.1 | 1:1 |
| CyPet-YPet | 477 | 530 | 0.51 | 104,000 | 5.1 | 1:4.5 |
| EGFP-mCherry | 507 | 610 | 0.60 | 72,000 | 5.1 | 2.5:1 |
| Venus-mCherry | 528 | 610 | 0.57 | 72,000 | 5.7 | 3:1 |
| Venus-tdTomato | 528 | 581 | 0.57 | 138,000 | 5.9 | 1:2 |
| Venus-mPlum | 528 | 649 | 0.57 | 41,000 | 5.2 | 13:1 |
目前对绿色等级(500纳米至525纳米)的FRET报告基因进行活细胞成像的最佳选择是GFP衍生物祖母绿,其特性与其EGFP母体相似。祖母绿包含 EGFP 中的F64L和S65T突变,但该变体还有四个额外的点突变,可改善折叠、37 摄氏度下的表达和亮度。最近,绿色光谱区域的新成员被创造出来,称为超级文件夹GFP,它比EGFP或祖母绿更亮,更耐酸,并且具有相似的光稳定性。因此,超级文件夹变体应该是与哺乳动物蛋白质融合和构建FRET生物传感器的绝佳候选者,特别是那些与标准GFP衍生物表现出折叠问题的传感器。另一种明亮的荧光报告基因,可能是一个很好的FRET候选者,被称为Azami Green,已经从石珊瑚Galaxeidae中分离出来,并被证明在哺乳动物细胞系中表达时迅速成熟。此外,已经报道了通过定点和随机诱变结合青色蛋白文库筛选获得的两种明亮的单体GFP报告基因。mWasabi源自Clavularia珊瑚属,是蓝色荧光蛋白的潜在替代绿色发射FRET伙伴,因为在400纳米和更低的蓝色变体经常被激发时吸光度可以忽略不计。新的绿色报告基因是市售的(等位基因生物技术),对于与长斯托克斯位移蛋白(如T-蓝宝石)以及与靶向蛋白融合中的定位标签的双色成像特别有用。TagCFP的衍生物,称为TagGFP,是一种明亮的单体绿色变体,在482纳米处具有最大吸收,在505纳米处发射。TagGFP仅比EGFP略亮,可作为Evrogen的克隆载体和融合标签使用,但在文献报告中尚未得到彻底表征。
黄色荧光蛋白(525纳米至555纳米)是迄今为止开发的最通用的基因编码探针之一,应该提供在FRET配对中充当供体和受体的候选物。被称为黄水晶和金星的变体是目前该光谱类别中最有用的蛋白质(见表1),但两者都没有商业上可用。另一种变体以生日石黄玉命名,可从 Invitrogen 获得,并已在融合标签定位、细胞内信号传导和 FRET 研究中发挥作用。Evrogen “Tag”商业化系列定位报告蛋白的新成员TagYFP是一种单体水母(Aequorea macrodactyla)衍生物,其亮度略低于EYFP,但可光性高一个数量级。与其合作伙伴类似,TagYFP(524纳米的发射峰)尚未在文献中表征,但可以作为哺乳动物克隆载体或融合标签购买。
在导致CyPet产生的相同荧光激活细胞分选研究中(如上所述),还获得了进化优化的互补FRET受体,称为YPet。YPet以其对FRET(YFP代表energytransfer)的熟练程度命名,是迄今为止开发的最亮的黄色变体,并表现出合理的光稳定性。YPet提供的对酸性环境的抵抗力优于Venus和其他YFP衍生物,这将增强该探针在针对酸性细胞器的生物传感器组合中的实用性。然而,尽管优化的CyPet-YPet组合应该是开发新型FRET生物传感器的首选起点,但YPet性能提高的起源仍然存在严重疑问,这可能仅仅是由于与其共同进化的合作伙伴CyPet的二聚化增强。同样,CyPet和YPet在融合标签中对定位实验、双分子互补分析和其他应用的适用性尚未确定。这两种蛋白质都以弱二聚体的形式存在于溶液中,但可能可以使用与其他Aequorea变体配合良好的A206K突变转化为真正的单体(尽管这显然破坏了它们在FRET中的优势)。
橙色荧光蛋白,所有这些都是从珊瑚礁物种中分离出来的,有可能在各种FRET成像场景中有用。也许其中最通用的是单体Kusabira Orange,这是一种最初从蘑菇珊瑚Fungia concinna(日语称为Kusabira-Ishi)中提取的蛋白质。Kusabira Orange(缩写为mKO)的单体版本是通过通过定点和随机诱变引入20多个突变而创建的。该单体(可从MBL国际公司获得)具有与四聚体相似的光谱特性,并且具有类似于EGFP的亮度值,但对酸性环境比其母体略微敏感。然而,该报告基因的光稳定性是所有光谱类别中最好的蛋白质之一,使mKO成为长期成像实验的绝佳选择。此外,发射光谱图与青色荧光蛋白充分分离,以提高包含mKO的生物传感器的FRET效率,并且该探针可用于使用青色,绿色,黄色和红色探针组合的多色研究。
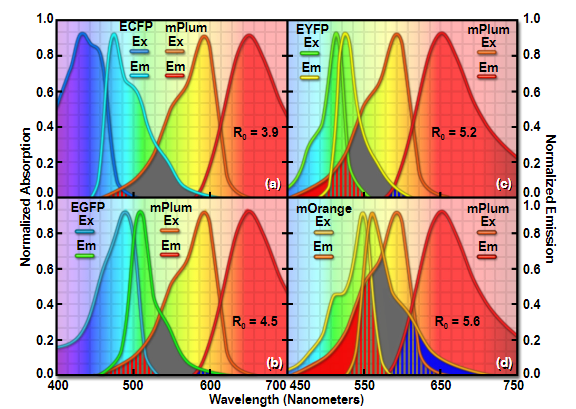
图 10-荧光蛋白 FRET 与远红受体的配对
图10所示是ECFP(图10(a))、EGFP(图10(b))、EYFP(图10(c))和mOrange(图10(d))的光谱图,每个都充当mPlum的FRET供体,mPlum是一种远红发射荧光蛋白受体。随着供体的发射光谱分布向更长的波长(从青色到橙色),光谱重叠(填充灰色区域)和计算的Förster距离(R(0))相应增加。类似地,激发和发射串扰(分别为红色和蓝色阴影区域)也随着供体和受体发射峰之间的波长分离减小而增加。请注意,ECFP和mPlum在激发光谱中仅表现出有限程度的重叠,而在发射光谱中几乎没有重叠。相比之下,当mOrange与mPlum配对时,激发和发射串扰水平都很高。随着荧光蛋白调色板的不断扩展,研究人员应该很容易获得广泛的新FRET对。
mRFP1衍生物mOrange比mKO略亮,但光稳定性不到10%,因此严重损害了其在需要重复成像的实验中的应用。然而,mOrange仍然是橙色光谱类别中最亮的蛋白质之一,并且在强度比长期光稳定性更关键的情况下仍然是一个很好的选择。此外,结合绿色发射的T-Sapphire,mOrange是CFP-YFP蛋白的合适替代品,作为FRET对产生更长波长的生物传感器,并且可以与其他光谱区域的蛋白质偶联以进行多色研究。mOrange的改进版本(名为mOrange2)具有显着提高的光稳定性。最近,一种名为TagRFP的明亮的新单体橙色蛋白被引入作为定位和FRET研究的候选物,并可能被证明在多种生物传感器结构中有效。任何光谱类别中最亮的荧光蛋白是二聚体番茄(dTomato)的串联版本,这是一种橙色衍生物,是原始水果蛋白之一。番茄蛋白在N端和C端含有来自GFP的第一个和最后七个氨基酸,以增加对融合蛋白的耐受性,减少定位中的潜在伪影,并增强其在FRET生物传感器中的使用可能性。串联二聚体版本(实际上是单体)是通过将dTomato的两个拷贝从头到尾与23个氨基酸接头融合而创建的。由于双发色团的存在,所得的tdTomato非常明亮,并具有出色的光稳定性。使用这种蛋白质的主要缺点是尺寸较大(是单体蛋白质的两倍),这可能会干扰某些生物聚合物中的融合蛋白包装。
寻找理想的红光荧光蛋白一直是使用FRET生物传感器和融合进行活细胞和全动物成像的目标,这主要是由于在多色成像实验中需要该光谱区域的探针,以及更长的激发波长产生较少的光毒性并且可以更深入地探测生物组织。迄今为止,已经报道了广泛的潜在有用的红色探针(590纳米至650纳米的发射),其中许多仍然受到其原产物种赋予的强制性四级结构的某种程度的影响。与水母蛋白不同,珊瑚礁蛋白的大多数天然和基因工程变体在37摄氏度下非常有效地成熟。广泛的诱变研究工作,包括新引入的方法,已成功应用于寻找黄色、橙色、红色和远红色荧光蛋白变体,进一步降低这些潜在有效的生物探针自我结合的趋势,同时将发射最大值推向更长的波长。结果是改进的单体蛋白具有增加的消光系数、量子产率和光稳定性,尽管还没有单一变体通过所有标准进行优化。
红色mFruit蛋白mApple,mCherry和mStrawberry(发射峰值分别为592,610和596纳米)的亮度水平范围为EGFP的50%至110%,但mApple和mCherry的光稳定性远高于mStrawberry,是替代mRFP1进行长期成像实验的最佳探针选择。通过迭代体细胞超突变进一步扩展mFruit蛋白光谱类别,产生了两种新的荧光蛋白,其发射波长最大值为625和649纳米,代表了第一个真正的远红基因工程探针。这对探针中最可能有用的探针被命名为mPlum,它的亮度值相当有限(EGFP的10%),但具有出色的光稳定性。这种单体探针应该与在青色、绿色、黄色和橙色区域发射的变体结合使用,用于多色成像实验,并作为绿色和黄色蛋白质(如祖母绿和黄水晶)的生物传感器FRET伙伴(见图10)。已经从珊瑚礁生物中分离出几种显示出不同程度前景的红色荧光蛋白。将位点特异性和随机诱变应用于TurboRFP变体,然后筛选表现出远红荧光的突变,产生了一种名为Katushka的二聚体蛋白(发射最大值为635纳米)。虽然只有EGFP的三分之二亮度,但Katushka在光谱窗口中表现出最高的亮度水平,包括650至800纳米,这是一个对深层组织成像很重要的区域。将四种主要的Katushka突变引入TagRFP中产生了一种名为mKate的单体远红蛋白,具有相似的光谱特征。据报道,mKate的光稳定性非常出色,并且该蛋白质显示出与mCherry相似的亮度,这使其成为光谱远红部分定位和FRET实验的绝佳候选者。
尽管在将荧光调色板扩展到光谱的橙色、红色和远红色区域方面取得了重大进展,但青色和黄色Aequorea衍生物仍然是开发有用生物传感器的最有用的配对方案。这种不可预见的差异之所以发生,是因为大多数橙色和红色珊瑚衍生的蛋白质表现出相对宽的吸收光谱图,具有延伸到紫色和青色区域的长激发尾,从而产生直接的受体激发。另一个可能起作用的因素是融合荧光蛋白伴侣的相对成熟率。在大多数情况下,来源于Aequorea蛋白的变体比从珊瑚礁获得的变体成熟得更快,因此未成熟的受体可能导致许多珊瑚衍生蛋白质表现出的致敏化不良。此外,橙色和红色蛋白质的宽吸附光谱,加上单体版本的量子产率降低,可能使它们难以用于FRET。荧光蛋白FRET实验设计的未来成功将集中在匹配配对蛋白的成熟率等因素上。
结论
尽管基于无处不在的荧光蛋白家族的FRET实验为揭示活细胞系统中的分子动力学提供了巨大的潜力,但目前还没有完美的FRET对。CFP和YFP的优化版本仍然为一般用途提供了最有效的配对,尽管更好的组合可能迫在眉睫。同样,没有完美的技术来测量FRET,尽管上述方法都具有可以根据所研究的特定实验情况利用的优势。随着更多优化的荧光蛋白的出现,包括可能适合作为GFP或YFP供体受体的鲜红色变体,使用荧光蛋白的FRET应该在活细胞中的蛋白质 - 蛋白质相互作用研究中变得更加有用。如前所述,当前红色荧光蛋白调色板的宽吸收光谱,以及单体版本的较低量子产率,使得这些候选物难以在FRET中使用。然而,荧光蛋白改进的快速步伐使人们乐观地认为,这些蛋白质将在不久的将来可用,并将有助于进一步彻底改变这种阐明细胞内生化机制的新方法。

